Epilepsy Genetics
Epilepsy is a diverse group of conditions and syndromes that is characterized by recurrent, unprovoked seizures. Epilepsy affects about 1% of the population, including 1 out of 200 children. Over one-third of patients with epilepsy experience intractable seizures that do not respond to anti-seizure medications. In children with epilepsy, seizures are often accompanied by neurodevelopmental comorbidities, such as intellectual disability, autism spectrum disorder, behavioral disorders, and schizophrenia. In addition, the risk of death in is higher in patients with epilepsy both from accidents related to seizures as well as Sudden Unexpected Death in Epilepsy (SUDEP), which has been noted to be associated with a growing number of epilepsy-related genes.
As a result of rapid developments in next-generation sequencing and other genetic technologies, over 2,000 genes are now associated with epilepsy in the literature. Particularly in childhood-onset epilepsy, the genetics of epilepsy are complex, and targeted treatments remain elusive for the majority of patients. Pathogenic variants (disease-causing DNA variants) can be inherited from one or both parents, or they can arise de novo during embryogenesis. Some forms of epilepsy, often severe early-onset epilepsies, can be caused by a pathogenic variant in a single gene, the focus of gene discovery efforts to date. In contrast, some more common forms of epilepsy may be influenced by combinations of pathogenic variants in several genes, which the field has yet been powered to study. Numerous different molecular pathways are implicated in epilepsy so far, including ion channels, receptor subunits, and cell-signaling molecules. Our research group is actively involved in both epilepsy gene discovery and genotype-phenotype studies to elucidate the complex nature of the genetic epilepsies and to inform functional models of genetic epilepsies.
Click the buttons below to learn more about our epilepsy research interests.


Classical seizure observed in a 5dpf larval zebrafish: Stage 1 – increase in swimming velocity, Stage 2 – whirlpool-like movements, Stage 3 – loss of posture.
Functional Models of Epilepsy in Zebrafish
Zebrafish (Danio rerio) represent an extremely versatile model system for studying genetic disease and neurodevelopment. Their high genetic homology to humans (75-85%), ease of genetic manipulation using CRISPR/Cas9 genome editing, rapid generation times, and large clutch sizes are all advantages in comparison to other vertebrate models. Neural development occurs rapidly and can be readily observed using fluorescent markers in translucent zebrafish. Observable seizure-like behaviors as well electrophysiological responses to pro-convulsant drugs have been recorded in larval zebrafish, making them a useful model for epilepsy. Due their small size, high-throughput screens of novel chemical compounds can be readily performed on larval zebrafish to identify molecular targets for further experimental investigation, with the ultimate goal of conducting clinical drug trials that can bring targeted treatments to human patients.

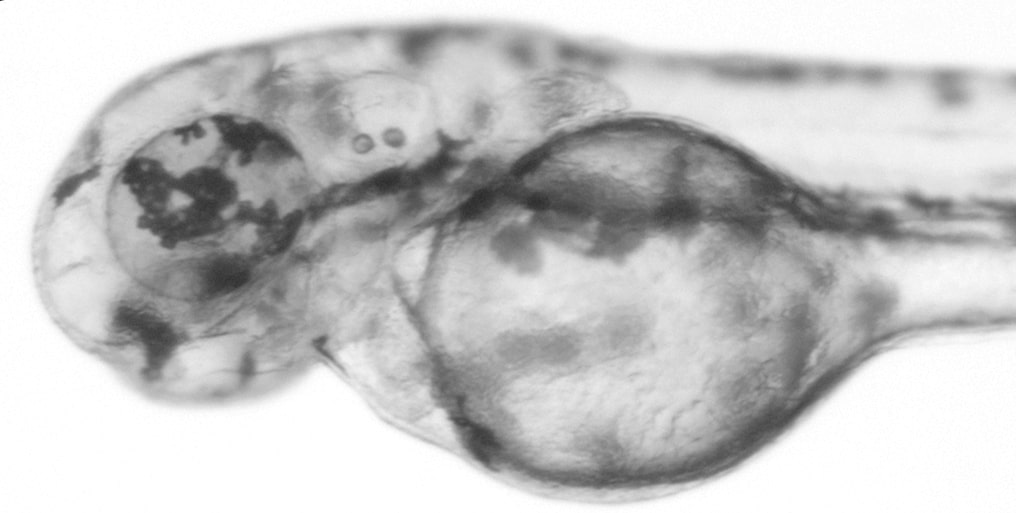
This zebrafish larva was was injected with slc24a5 CRISPR sgRNA and Cas9 protein at the 1-cell stage. Expression of slc24a5 is required for retinal pigment epithelium development. In the CRISPR-injected embryo, there is mosaic depigmentation in the eye due to the introduced mutations in slc24a5. Image courtesy of Jeremy Ullmann.
Zebrafish Models of Candidate Epilepsy Genes
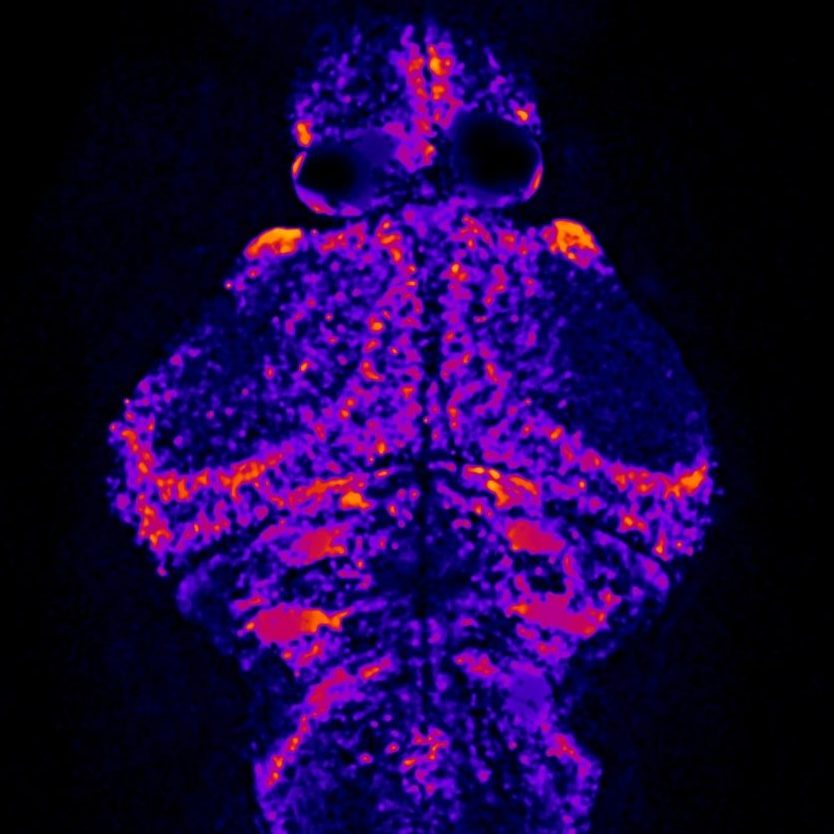
Our laboratory is modeling candidate epilepsy genes from the literature as well as our own candidates from sequencing projects in our Epilepsy Genetics Programs. Using CRISPR/Cas9, we create knock-out zebrafish models of the most promising candidate genes. Then, we evaluate these transgenic zebrafish lines for spontaneous behavioral seizures as well as increased seizure susceptibility in response to pro-convulsant drugs, hyperthermia, or certain frequencies of light flashes. We have developed the ability to record local field potentials from the telencephalon or optic tectum of larval zebrafish to observe for electrophysiological changes or to use micro-electrode array (MEA) recordings to do the same, analogous to an electroencephalogram (EEG) in humans.
In addition to our zebrafish models, we have partnered with the Translational Neuroscience Center, Harvard Stem Cell Institute, and Woolf Lab to conduct studies of neurons derived from patient cell lines representing the genes SCN8A and KCNQ2. Our emerging pilot studies seek to demonstrate the electrophysiological abnormalities that can be detected in this system and to screen abnormal lines with compound libraries in an attempt to discover novel treatments for patients with pathogenic variants in these genes.
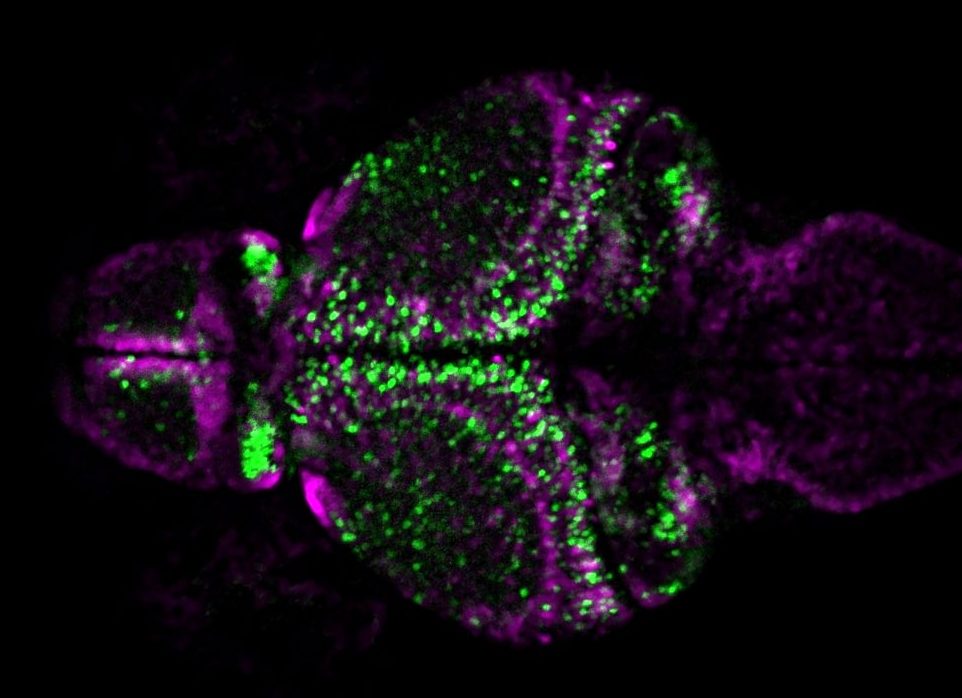
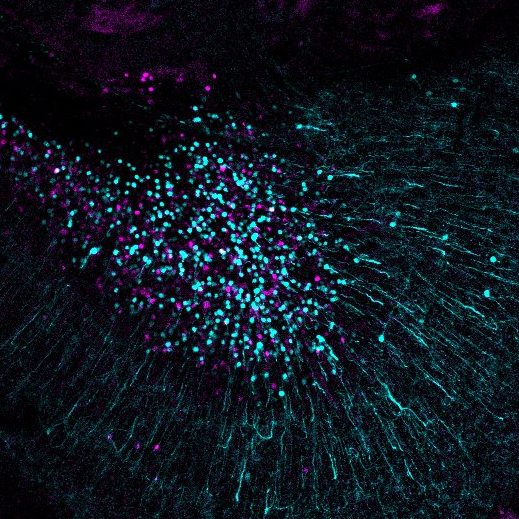
Larval zebrafish brain with inhibitory neurons labelled with magenta and excitatory neurons labelled with green. Image courtesy of Barbara Robens.
Zebrafish Models of PCDH19-Related Epilepsy
Pathogenic variants in PCDH19 are associated with X-linked “female-limited epilepsy.” DNA variants can occur de novo or can be inherited from mildly affected or unaffected mothers or, interestingly and unlike most X-linked diseases, from unaffected fathers. It is hypothesized that obligate mosaicism is responsible for limiting the disease almost exclusively to females, whereas it is rarely seen in males unless also in the mosaic state. The wide range of phenotypes seen in this disorder can include refractory childhood-onset seizures, intellectual disability, and autism.
In cooperation with the Epilepsy Genetics Clinic and our PCDH19 Registry, which currently has >50 patients registered and continues to recruit patients and their families from around the world, we aim to characterize the pathogenic variants of PCDH19 and establish genotype-phenotype correlations in these patients as well as to inform our zebrafish studies of this gene.

Tanks of adult zebrafish at Boston Children’s Hospital. Photo Courtesy of Chris Lawrence.
Zebrafish Models of Epilepsy and Schizophrenia
Traditionally, epilepsy and schizophrenia have been thought of as two separate neurological conditions, but evidence for a shared biological basis has recently emerged. Individuals with epilepsy and more likely to develop schizophrenia, and vice versa. Moreover, the two disorders have been found to share some genes as their bases. Our lab has partnered with the Schier lab to model genes associated with both diseases using transgenic zebrafish, assess the models for features relevant to both diseases, and then prioritize genes for high-throughput chemical compound screens in zebrafish to identify potential therapeutic drug compounds for pre-clinical trials.
Human Genetics
Our functional studies in zebrafish and other model systems are all closely informed by the ongoing human genetics research performed by our clinical research team members. We have projects ranging from all stages of translational medicine, from gene discovery to genotype-phenotype and natural history studies to clinical trials.


Gene Discovery Projects in Epilepsy
Our early efforts in gene discovery focused on somatic mosaic, post-zygotic mutations in focal epilepsy related to developmental brain malformations. We continue these efforts in partnership with the Core for Neurological Diseases and the group of Dr. Erin Heinzen (University of North Carolina), with single cell sequencing to be performed on mutation-positive cases in the laboratory of Dr. Christopher Walsh (BCH).
Through our own lab’s studies as well as work with the Epilepsy Phenome/Genome Project and Epi4K, both international NIH-supported epilepsy genetics consortia, we have discovered numerous de novo single-gene causes of severe early onset epilepsies that point not only to dysfunction or disruption of ion channels but also several other processes, including synapse development and early brain organization. We continue to apply cutting-edge sequencing and analysis methods to the many cases that remain unsolved.
A major effort beginning in 2018 has been the BCH Genomics Pilot, through which we are enrolling and sequencing over 500 trios (patients and parents). The resulting 3000+ exomes are analyzed using the BCH WuXi NextCode platform, and our team then designates which findings merit clinical confirmation and return of results to families as well as which genes merit additional study in our bench laboratory.
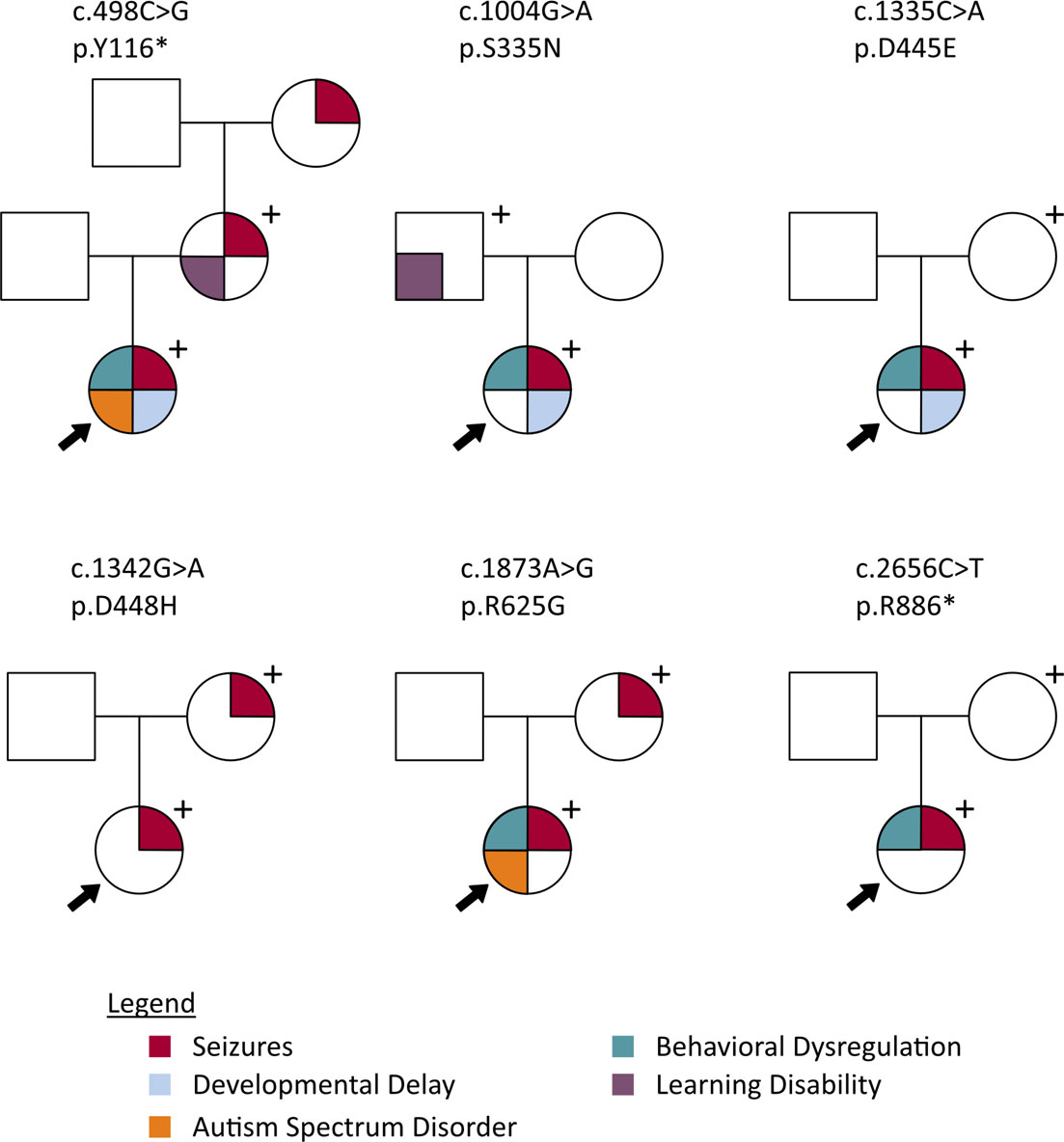
Pedigrees displaying phenotypes of probands and genotype-positive parents for 5 different PCDH19 variants. From Smith et al. 2018, Epilepsia.
Genotype-Phenotype and Natural History Studies
We have engaged with parent-led foundations to create multi-center registries for patients with pathogenic variants in genes such as PCDH19 and KCNQ2.
The PCDH19 Registry was launched in 2014 in collaboration with the University of California, San Francisco. The Registry collects clinical and genetic information on individuals with PCDH19 variants to be used for future research. We currently have 75 participants, plus additional genotype-positive family members, enrolled. In addition, we are coordinating formal neurodevelopmental evaluations of children with PCHD19-related epilepsy to better characterize the cognitive and behavioral profile of this population.
In collaboration with the KCNQ2 Alliance, we are collecting phenotypic information on individuals diagnosed with KCNQ2-related disorders through a complex questionnaire focused on symptoms and outcomes. We hope to use this natural history study as a foundation for larger-scale efforts to identify targeted therapies for this unique patient population. We have currently enrolled approximately 70 individuals in this study.
Additionally, we actively participate in a consortium with Emory (Traynelis Lab), Denver Children’s (Benke Lab), and CHOP and contribute cases for phenotypic study and functional evaluation through the Emory Center for Functional Evaluation of Rare Variants (GRIN2A, GRIN2B, GRIN2D, GRIA, GRIK, and GRID).
Clinical Trials
We have conducted gene-specific industry-sponsored clinical trials for patients with SCN1A– and PCDH19-related epilepsies. We anticipate that trials will continue as our functional studies in models of genetic epilepsy lead us toward rational and precision treatments for children with severe epilepsy.
We have designed n of 1 trials as appropriate for genetic epilepsies and obtained permission from our IRB and the FDA to administer investigational new drugs for patients with epilepsy due to variants in SCN1A, GRIN2A, SYNGAP1, CAD, and PLCB1 (collaboration with Dr. Gerard Berry, Division of Genetics).
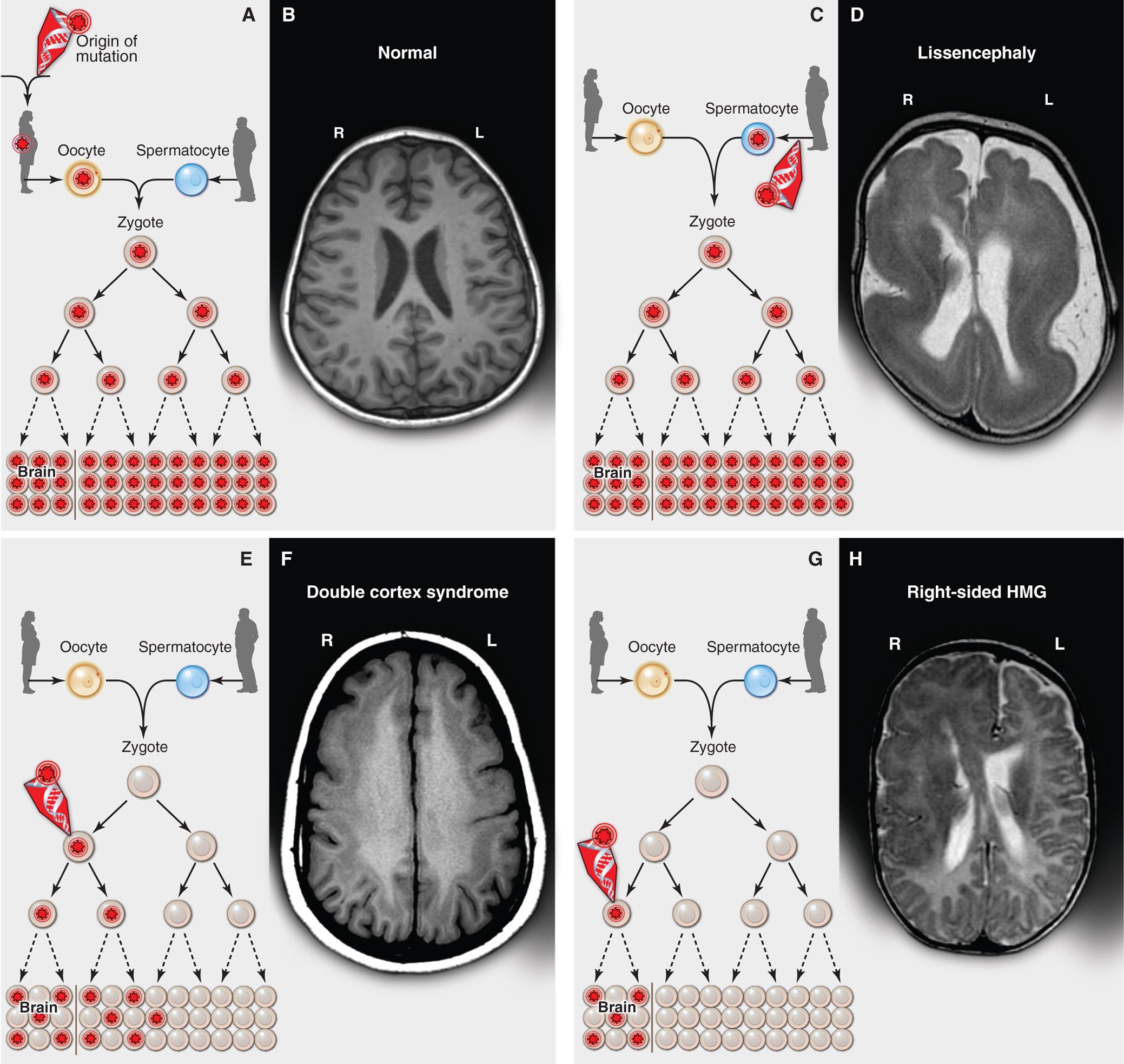
Inherited, de novo, and somatic mutations causing neurological disease. From Poduri et al. 2013, Science.
Gene and Variant Curation for Brain Malformations
The ClinGen Brain Malformation Expert Panel, chaired by Dr. Poduri and Dr. Timothy Yu (BCH), was formed to curate genes and variants associated with developmental brain malformations. Somatic DNA variants arising during embryogenesis and fetal development are increasingly recognized as important causes of human diseases, now extending beyond just cancer into the realm of developmental disorders and epilepsy. Post-zygotic DNA variants in the mTOR pathway (including MTOR, AKT3, PIK3CA, PIK3R2, and DEPDC5) are known to cause a spectrum of brain overgrowth syndromes ranging from focal cortical dysplasia to dysplastic megalencephaly. Proper interpretation of somatic DNA variants requires consideration of several factors not accounted for in the current guidelines which focus predominantly on germline variation. Our group has drafted a framework for the curation of somatic mosaic (post-zygotic) variants that addresses many of the issues unique to somatic variants.
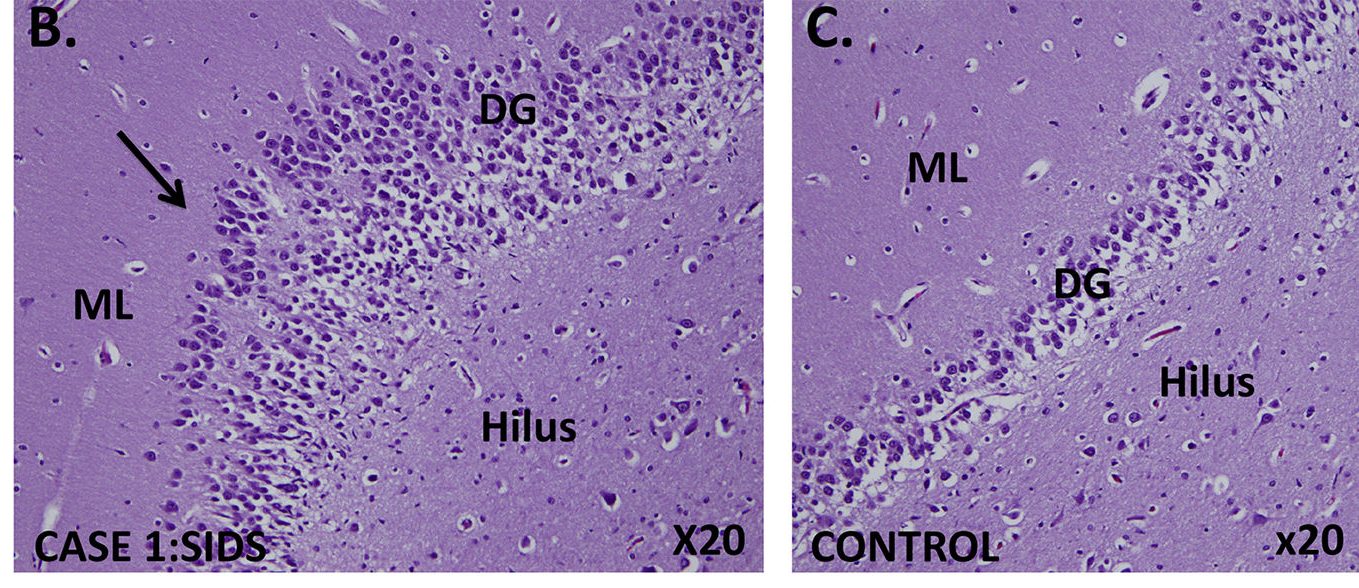
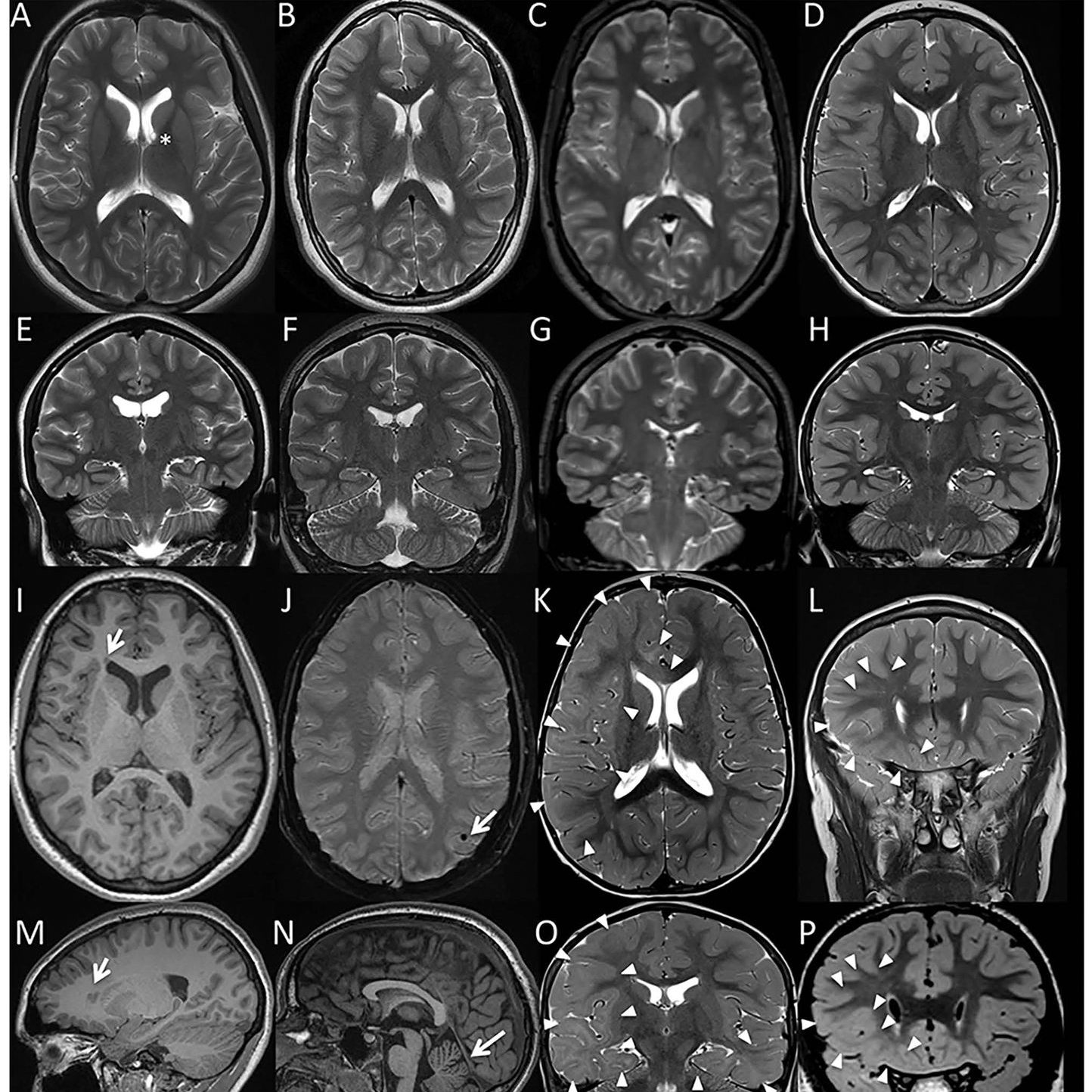
Abnormal developmental lesions in the dentate gyrus region of the hippocampus were identified in two infants with SIDS and variants in SCN1A. As shown in the case on the left, focal bilamination and other cellular abnormalities are visible in the dentate gyrus. An age-matched control infant (on the right) shows the normal single layer of dentate gyrus granule cells. From Brownstein et al. 2018, Epilepsia.
Collaborative Studies on SIDS and Sudden Death in Childhood
Through a collaboration with Robert’s Program on Sudden Death in Childhood, we are investigating epilepsy-related neuropathological findings in infants and children who die suddenly without a history of epilepsy. We are taking an undiagnosed disease approach to individual cases, and an ‘epilepsy genetics’ and somatic variant approach to cases for whom DNA is available.
For more information on studies or trials for patients with genetic epilepsies, please contact the Epilepsy Genetics Program.