The Engle Lab
at the F.M. Kirby Neurobiology Center
Science
For a full list of publications, please click here. To request a PDF of a lab publication, email engle.admin@childrens.harvard.edu.
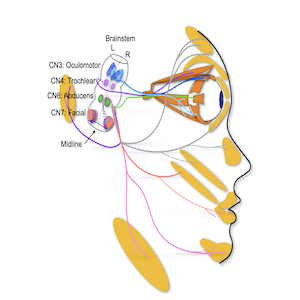
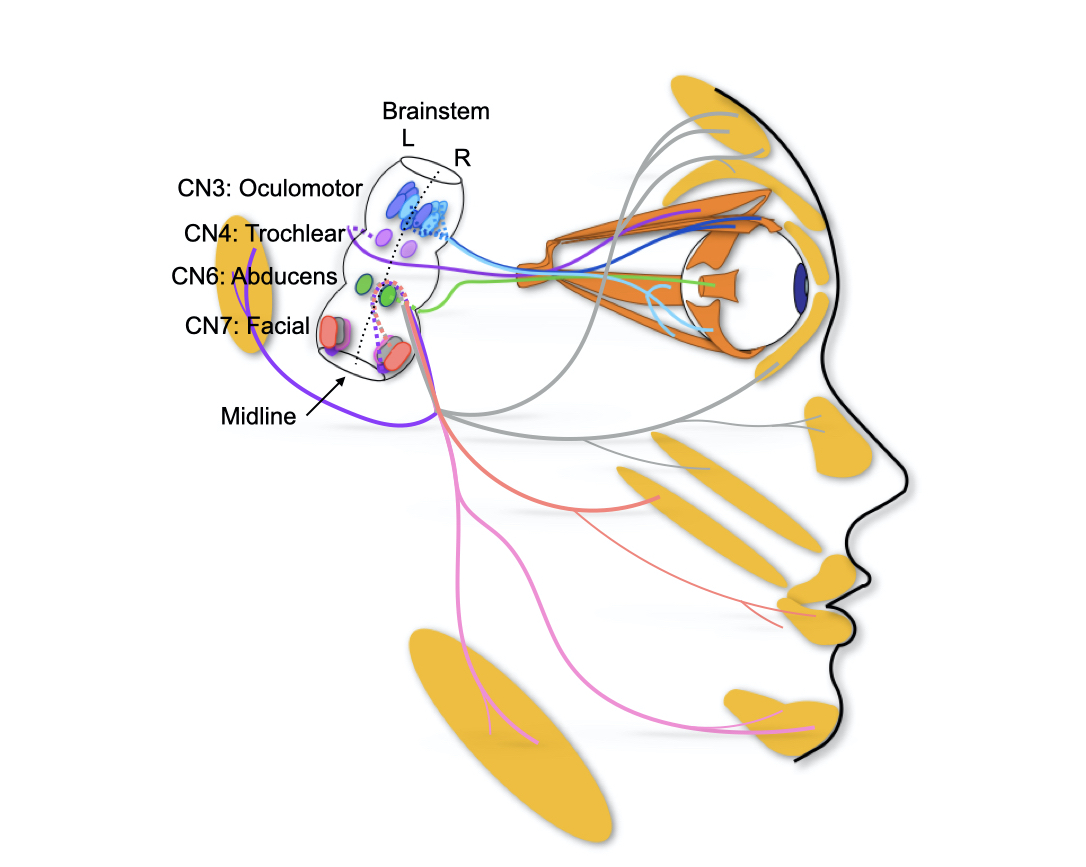
Lateral view of human brainstem and face highlighting a subset of cranial motor nuclei and nerves. Eye and face movements are accomplished through four of our twelve cranial nerves (CN). Oculomotor (CN3), trochlear (CN4) and abducens (CN6) innervate the extraocular muscles while facial (CN7) innervates the facial muscles.
Publications
1. Gutowski NJ, Bosley TM, Engle EC. 110th ENMC International Workshop: the congenital cranial dysinnervation disorders (CCDDs). Naarden, The Netherlands, 25-27 October, 2002. Neuromuscul Disord. 2003 Sep;13(7-8):573-8. PMID: 12921795 >
2. Engle EC*. Human genetic disorders of axon guidance. Cold Spring Harbor Perspect. Biol. 2010 Mar;2(3):a001784. PMCID: PMC2829956 >
3. Whitman MC, Engle EC*. Ocular Congenital Cranial Dysinnervation Disorders (CCDDs): Insights into Axon Growth and Guidance. Hum Mol Genet. 2017 Aug 1;26(R1):R37-R44. PMCID: PMC5886468 >
Congenital Cranial Dysinnervation Disorders (CCDDs)
We humans are defined, in part, by the nuanced and coordinated ocular and facial movements that are fundamental to our vision and social communication. The Engle Lab studies human developmental disorders that perturb these eye and face movements as a framework to understand how neurons are specified and how cranial neural circuits are formed. We identify human disorders of eye and face movement, define their genetic etiologies, and study the developmental pathology and disease mechanisms in vitro and in animal models. We also study the normal development of cranial motor neurons and their axonal processes.
Our research has led to the definition of the congenital cranial dysinnervation disorders (CCDDs). The CCDDs are birth defects in which cranial motor neurons or their axonal processes fail to develop correctly. While the CCDDs include all cranial motor nerves, the lab focuses in particular on disorders of oculomotor (cranial nerve 3), trochlear (cranial nerve 4), abducens (cranial nerve 6) and/or facial (cranial nerve 7) development. The ocular CCDDs result in the inability to raise the eyelid(s) or to move the eye(s) in one or more directions of gaze. The facial CCDDs result in unilateral or bilateral congenital facial weakness. We also study the genetics of esotropia and exotropia (common or comitant forms of strabismus), as described below.
The lab has found that human disorders of cranial motor neuron development provide a unique and powerful system to study Mendelian genetics and neural connectivity:
- Each cranial motor nucleus contains a small number of motor neurons.
- Each cranial motor nerve has straightforward tractable developmental anatomy that can be visualized in three dimensions.
- Errors in development result in human phenotypes that are present at birth, lifelong, visible to the examiner, and stereotypic in presentation.
- All higher vertebrates have 12 highly conserved cranial nerves, providing consistent translation to model organisms for developmental and mechanistic studies.
- The Engle Lab has amassed a remarkable Mendelian cohort of research participants, leading to a high likelihood that we will identify multiple disease alleles with which to document causality of new genetic disorders.

Figure 1. Small sample of CCDD pedigrees from the Engle Lab whole genome sequence cohort. AD = autosomal dominant; AR = autosomal recessive. Filled symbol = affected, central dot = obligate carrier, suggesting incomplete penetrance.

Figure 2. Validation of a pathogenic deletion ascertained by whole exome/whole genome sequencing in a patient with horizontal gaze palsy. Modified from Dobyns et al., AJHG, 2018, PMCID: PMCID: PMC6288423 >

Figure 3. CCDD caused by complex structural rearrangement. (Left) Structural variant and novel DNA-DNA interactions within a region of linkage visualized by HiC chromosome conformation capture. Capture HiC of affected minus unaffected family member reveals gain (red) and loss (blue) of DNA-DNA interactions in affected individual. (Right) Bulk RNA sequencing of lymphoblastoid cell lines from family members reveals a novel fusion protein in affected members that is absent in unaffected family members. Unpublished data.
Genetics & Genomics of CCDDs
The Engle Lab identifies novel genetic etiologies of heritable pediatric neurodevelopmental disorders, with a focus on brainstem malformation syndromes and congenital disorders of eye and face movement. We are particularly interested in phenotypes arising from errors in cranial motor neuron and axon development, collectively known as the congenital cranial dysinnervation disorders (CCDDs). We have recruited a Mendelian disease cohort of ~15,000 individuals, and their phenotypic analysis has led to the discovery or elaboration of multiple new congenital syndromes. We are coupling this phenotypic database with over 1000 exome (WES) and 1000 genome (WGS) sequences. These datasets provide ample opportunities for novel discovery and have led to the identification of myriad candidate variants in genes involved in neuronal differentiation and/or axon growth and guidance, as well as in conserved noncoding gene regulatory elements. The lab analyzes WES/WGS for coding and noncoding single nucleotide variants, small insertions and deletions, and larger coding and noncoding structural variants. To support pathogenicity, we then strive to identify additional alleles in candidate genetic loci. Subsequently, we conduct in vitro and in vivo studies to establish functional and mechanistic significance of the variant(s) and elucidate normal and abnormal function of the gene or noncoding sequence.
Publications
1. Miyake N, Chilton J², Psatha M², Cheng L, Andrews C, Chan W-M, Law K, Crosier M, Lindsay S, Cheung M, Allen J, Gutowski NJ, Ellard S, Young E, Iannaccone A, Appukuttan B, Stout JT, Christiansen S, Ciccarelli ML, Baldi A, Campioni M, Zenteno JC, Davenport D, Mariani LE, Sahin M, Guthrie S, Engle EC*. Human CHN1 mutations hyperactivate α2-chimaerin and cause Duane’s retraction syndrome. Science. 2008 Aug 8;321(5890):839-43. PMCID: PMC2593867 >
2. Tischfield MA, Baris HN², Wu C², Rudolph G, Van Maldergem L, He W, Chan W-M, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, Sabol LJ, Doherty EJ, de Uzcategui CE, de Uzcategui N, Collins ML, Sener EC, Wabbels B, Hellebrand H, Meitinger T, de Berardinis T, Magli A, Schiavi C, Pastore-Trossello M, Koc F, Wong AM, Levin AV, Geraghty MT, Descartes M, Flaherty MP, Jamieson RV, Moller HU, Meuthen I, Callen DF, Kerwin J, Lindsay S, Meindl A, Gupta ML*, Pellman D, and Engle EC*. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010 Jan 8;140(1):74-87. PMCID: PMC3164117 >
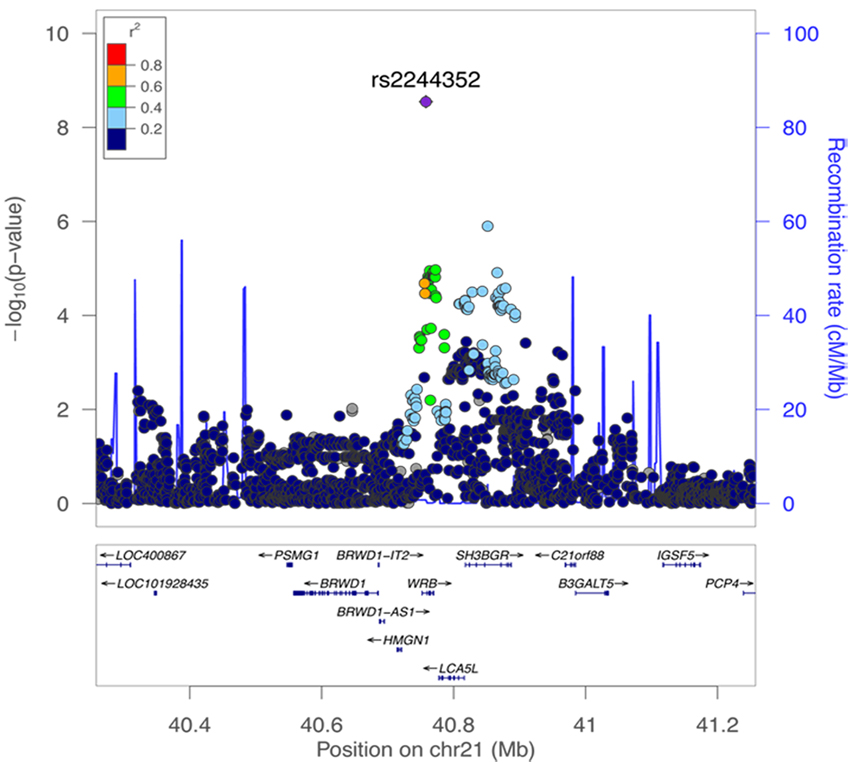
Regional association plot of a nonaccommodative esotropia discovery cohort based on a genome side association study (GWAS). The top-ranked SNP, rs2244352, is shown as a solid purple diamond. The color scheme indicates linkage disequilibrium between rs2244352 and other SNPs in the region using the r2 value calculated from the 1000 Genomes project. The y-axis is the -log10 (P value) computed for the displayed SNPs. From S Shaaban et al, IOVS, 2018, PMCID: PMC6088800 >
Genetics & Genomics of Esotropia & Exotropia
Esotropia and exotropia are common forms of comitant strabismus that affect 2-4% of the population. Unlike those with ocular CCDDs in which the eyes are restricted in their movement, individuals with esotropia or exotropia can move each eye fully but the eyes are misaligned, resulting in one eye turning inward (esotropia) or outward (exotropia). In most cases, the etiology of comitant strabismus remains unknown. While many families segregate these disorders with incomplete dominance, they appear to be oligogenic traits. The Engle Lab enrolls and phenotypes families that segregate esotropia or exotropia and studies them using a variety of genetic techniques, including linkage analysis, exome and genome sequencing, copy number variation analysis, and genome wide association studies (GWAS).
Publications
1. Shaaban S, MacKinnon S, Andrews C, Staffieri SE, Maconachie GD, Chan W-M, Whitman MC, Morton SU, Yazar S, MacGregor S, Elder JE, Traboulsi EI, Gottlob I, Hewitt AW, Strabismus Genetics Consortium, Hunter DG, Mackey DA, Engle EC*. Genome-wide association study identifies a susceptibility locus for comitant esotropia and suggests a parent-of-origin effect. Invest Ophthalmol Vis Sci. 2018 Aug 1;59(10):4054-4064. PMCID: PMC6088800 >
2. Whitman MC, Di Gioia SA, Chan W-M, Gelber A, Pratt BM, Bell JL, Collins TE, Knowles JA, Armoskus C, Pato M, Pato C, Shaaban S, Staffieri S, MacKinnon S, Maconachie GDE, Elder JE, Traboulsi EI, Strabismus Genetics Research Consortium, Gottlob I, Mackey DA, Hunter DG, Engle EC*. Recurrent Rare Copy Number Variants Increase Risk for Esotropia. Invest Ophthalmol Vis Sci. 2020 Aug 3;61(10):22. PMCID: PMC7443120 >
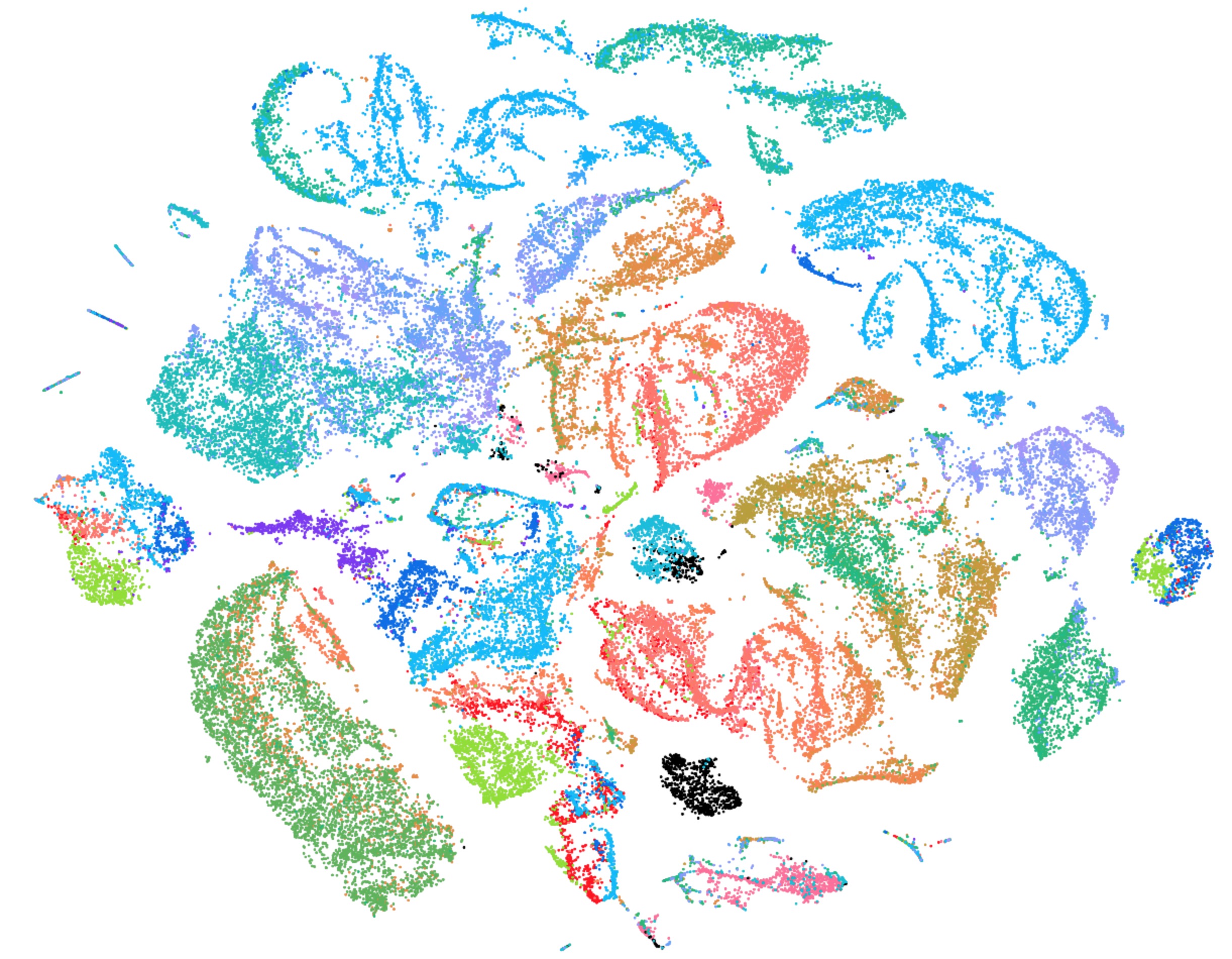
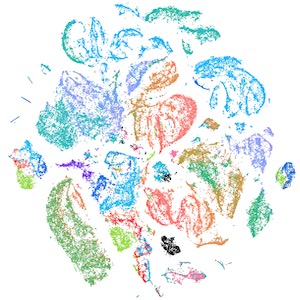
Figure 1. This tSNE shows single cell RNA-seq analysis of 70,000 mouse embryonic cells from the regions of CN3, CN4, CN6, and CN7 and enriched for motor neurons by fluorescent activated cell sorting (colored by sample ID). Unpublished data.

Figure 2. In addition to mutations in a gene’s UTRs or promotor, noncoding variants can alter long range cis-regulatory elements. These include enhancers that increase target gene expression and silencers that repress it. While cis-regulatory elements are extremely important for control of developmental gene expression, their role in Mendelian disorders is less well defined. We are actively studying the role of such elements in the CCDDs.
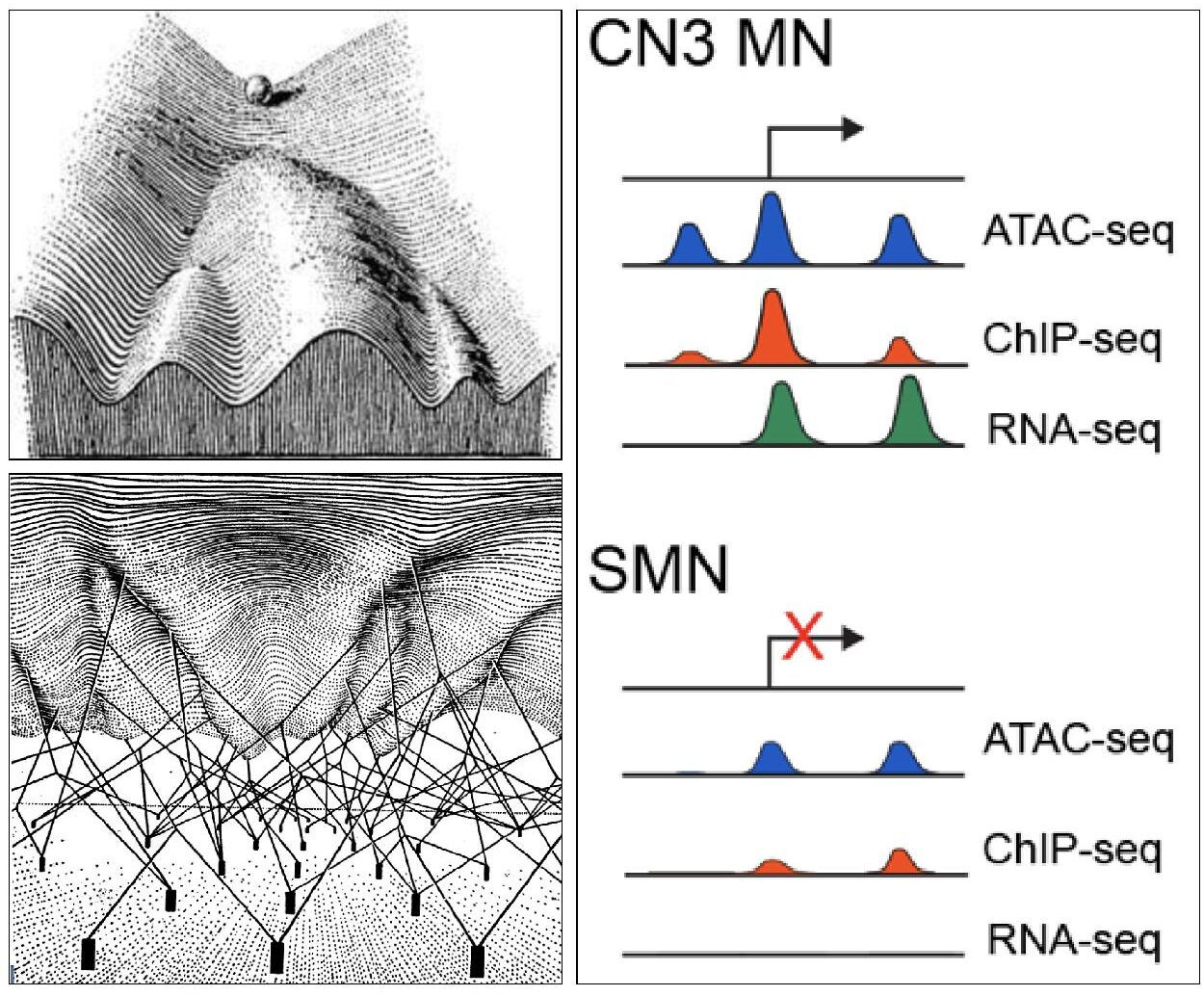
Figure 3. Developing cells negotiate a complex epigenomic landscape akin to a marble rolling down a hill (CH Waddington. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology. Allen & Unwin, London (1957)). The contours of the hill are moulded by “guy-wires” (transcription factors) that regulate gene expression through space and time. The Engle lab seeks to integrate multiple layers of epigenomic information in order to describe and predict the regulatory grammar of developing cranial motor neurons.
Transcriptomic and Epigenomic Analyses of the Developing Brainstem
The Engle Lab seeks to understand the signaling and gene regulatory events that underlie the formation of functioning cranial motor neurons and their circuits during embryonic development. These data inform neurodevelopment and provide a toolbox to study coding & noncoding Mendelian disorders.
Transcriptomic Profiling
We use an array of single cell RNA sequencing (scRNAseq) approaches to characterize the transcriptional identity of developing cranial motor nuclei. This has allowed us to identify and develop transgenic reporter mouse lines for specific cranial motor nuclei and projections, reagents that will help us characterize genetic models for human CCDDs. We also use these datasets as a springboard to investigate the early developmental steps that generate the functional diversity of cranial motor neurons.
Epigenomic Profiling
In order to understand regulation of cranial motor neuron and axon development, and to better interpret the rich catalogue of rare candidate variants in coding and non-coding space, the lab is generating cell type specific epigenomic profiles of developing cranial motor neurons at single-cell resolution. We then apply computational and experimental approaches to interweave chromatin accessibility, histone modification, and gene expression information from the cranial motor neurons to identify cell type specific developmental regulatory elements and their cognate genes. These data allow us to nominate causal regulatory mutations from participant genome sequencing data.
Movie 1: Movie of a 1TB lightsheet image of a cleared, intact developing embryonic day 10.5 mouse. This image shows the brainstem of an intact E10.5 Isl1-GFP+ mouse embryo stained for Isl1 protein (magenta, labels the motor neuron and some adjacent cell nuclei) and GFP protein (labels the motor neuron axons) and cleared with iDISCO+. It was imaged at 20X with a Zeiss Z1 Lightsheet at the Harvard Center for Biological Imaging and analyzed with Arivis. The various motor neuron (MN) populations are labeled in white (CN3, oculomotor; CN4, trochlear; CN5, trigeminal; CN6 abducens; CN7, facial; CN10, vagus; CN12, hypoglossal). Unpublished data.
Movie 2: Movie of an E15.5 cleared Isl-GFP mouse embryo in which one side has been imaged. Motor axons are labeled in green (immunostaining for GFP) and muscles and arteries are stained with anti-smooth muscle actin antibody. This movie shows the branches of the facial nerve. Unpublished data.
Movie 3: Movie of an E13.5 cleared Isl-GFP mouse embryo. Motor axons are labeled in green (immunostaining for GFP) and muscles and arteries are stained with anti-smooth muscle actin antibody. This movie shows the path of the ocular motor nerves (cranial nerves 3, 4 and 6) from the brainstem to the orbit. Unpublished data.
Imaging
We use a variety of imaging approaches to visualize development of the brain, cranial motor neurons and nerves. In humans, we study magnetic resonance and diffusion tensor imaging (MRI and DTI) of research subjects. In mice and zebrafish, we use advanced methods to clear embryonic brains for imaging.
We use lightsheet microscopy of cleared intact mouse embryos to visualize the motor neuron cell bodies and axons in the developing brainstem at high resolution. To do this we have combined tissue clearing and fluorescent labeling of whole organisms with advanced optics that can image through the entire mouse brainstem at 20X, achieving single cell resolution and visualization of multiple genetic markers. These approaches allow us to see the 3D spatial relationship of subpopulations within each motor nucleus (Movie 1).
To visualize the cranial nerves and the muscles they innervate, whole embryos or heads are immunostained and cleared using an iDisco protocol. Using confocal imaging, we create 3D reconstructions of the full path of the ocular motor axons from the brainstem to the extraocular muscles in the orbit, allowing us to assess if there are any defects in axon growth or guidance (Movies 2 and 3).
Publications
1. Whitman MC*, Nguyen EH, Bell JL, Tenney AP, Gelber A, Engle EC*. Loss of CXCR4/CXCL12 signaling causes oculomotor nerve misrouting and development of motor trigeminal to oculomotor synkinesis. Invest Ophthalmol Vis Sci. 2018 Oct 1;59(12):5201-5209. PMCID: PMC6204880 >
2. Grant PE*, Im K, Ahtam B, Laurentys CT, Chan W-M, Brainard M, Chew S, Drottar M, Robson CD, Drmic I, Engle EC*. Altered white matter organization in the TUBB3 E410K Syndrome. Cereb Cortex. 2019 Jul 22;29(8):3561-3576. PMCID: PMC6644882 >

Figure 1. Mouse model of KIF21A missense mutation resulting in CFEOM1. (A) 129/S1 Kif21a+/+ mouse with normal eyes. (B) 129/S1 Kif21aKI/KI mice with bilateral ptosis and globe retraction. (C, D) Confocal images of dissected and imaged proximal oculomotor nerves oriented with brainstem to the left and orbit to the right from E12.5 Kif21a+/+;IslMN:GFP (C) and Kif21aKI/KI;IslMN:GFP (D) embryos. Scale bar represents 100 mm. We found that the proximal mutant nerve was thickened and ended within the first half of the peripheral nerve trajectory as a single smooth, circumscribed enlargement that we refer to as the bulb. Distal to the bulb, each mutant nerve was thin. (E-H) We stained fluorescent OMN nerves with anti-neurofilament antibody to better visualize single axons. (E) and (F) correspond to regions of arrow and arrowhead in (C), while (G) and (H) correspond to regions of arrow and arrowhead in (D). Scale bars represent 20 mm. We found that Kif21aKI/KI axons proximal to the bulb had straight trajectories similar to WT. Within the bulb, a subset of axons pursued complex trajectories and appeared to terminate, while others maintained straight trajectories and exited the bulb to form the thin distal nerve. (I, J) Ultrastructural analysis of cross-section of E12.5 oculomotor wildtype nerves (I) consist primarily of axons running parallel to the nerve trajectory with a few central growth cones (star). By contrast, cross-section of mutant bulb (J) contain highly disorganized axons and increased numbers of longitudinal axons (arrows) as well as enlarged central growth cones, lamellipodia/filopodia, membranes and vesicles, and degenerating axons. Scale bars represent 500 nm. Through ex vivo anterograde and retrograde tracer studies we identified the axon population terminating within the bulb were primarily those destined to become the superior division of the oculomotor. Failure of their innervation of their target muscles results in ptosis and absent upgaze, the findings that define the human CFEOM1 phenotype. From L Cheng and J Desai et al, Neuron, 2014. PMCID: PMC4002761 >
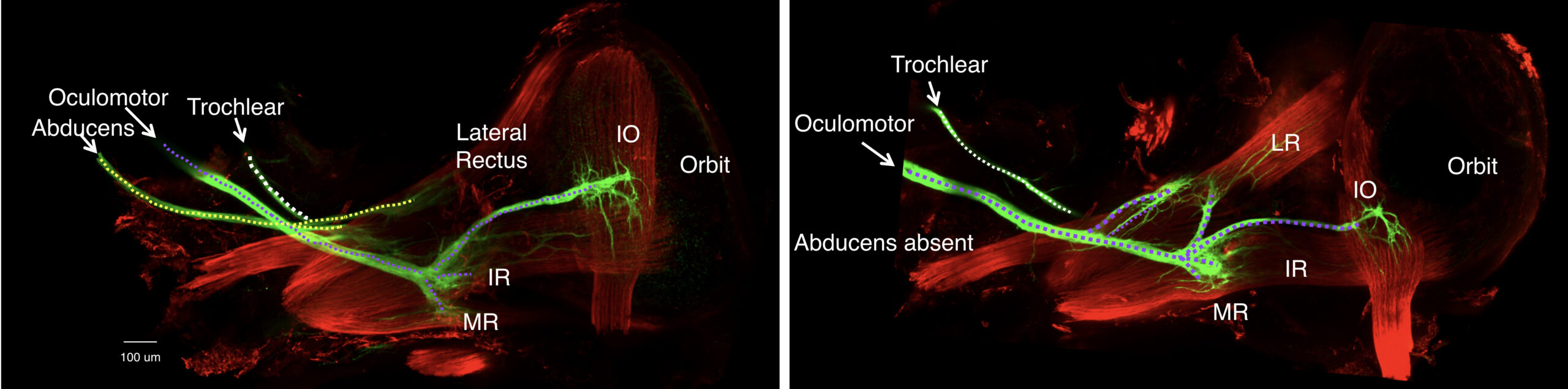
Figure 2. Duane retraction syndrome mouse model: abducens nerve is absent from the orbit and the oculomotor nerve misinnervates the lateral rectus muscle. Left: inferior view of extraocular muscle innervation in E16.5 wildtype mice. The abducens nerve (dotted yellow line) crossed the oculomotor nerve (dotted purple line) in close proximity to the lateral rectus muscle (LR) prior to its innervation. Right: inferior view of extraocular muscle innervation in E16.5 Chn1 KI/KI mice, a Duane syndrome mouse model. The abducens nerve is absent from the orbit. While the oculomotor nerve had a normal trajectory to the orbit, the lateral rectus is subsequently innervated by one or two aberrant oculomotor nerve branches arising at established oculomotor growth cone decision regions (purple dotted lines that extend to LR muscle): the proximal branch formed from the superior oculomotor nerve division branch-point off the main oculomotor trunk, while the distal branch arose from the inferior oculomotor nerve division branch-point to the medial rectus, inferior rectus, and inferior oblique muscles. We found similar misinnervation in the Mafb knockout mouse model of DRS. Thus, the oculomotor nerve provides compensatory misinnervation to the lateral rectus in the absence of the abducens nerve, confirming human autopsy and electromyographic data. From J Park, AMHG, 2016, PMCID: MC4908193 > and A Nugent, JCI, 2017, PMCID: PMC5409791 >
Figure 3. Microtubule association and motility of wildtype (left) and R954W mutant (right) full length Kif21a. (A, left) Single molecule imaging of mouse GFP-fused wildtype full length Kif21a proteins (green) moving on Cy5-labeled microtubules (red) in BRB12 motility buffer. Movie duration is 5 min. Note that only rare WT Kif21a are bound to microtubules and move processively, while a large number of motors remain in solution. (B, right) Single molecule imaging of mouse GFP-fused R954W mutant FL Kif21a proteins (green) moving on Cy5-labeled microtubules (red) in BRB12 motility buffer. Movie duration is 5 min. Note that compared to WT, many more mutant Kif21a proteins are bound to microtubules and move processively. From L Cheng and J Desai et al, Neuron, 2014. PMCID: PMC4002761 >
Figure 4. Tracking of axonal growth cones from E12.5 dorsal root ganglia (DRG) explant cultures on DIV3 from (left) wildtype and (right) Kif21a knockout mouse embryos by live imaging. Growth cone outgrowth was recorded every minute over 30 minutes using a Nikon Ti inverted microscope with PH1 filtered bright filled light. Images were analyzed using ImageJ software. Colored lines track growth cone trajectories over the 30-minute recording and revealed that the Kif21a knockout growth cones had less displacement and reduced forward growth. Unpublished data.
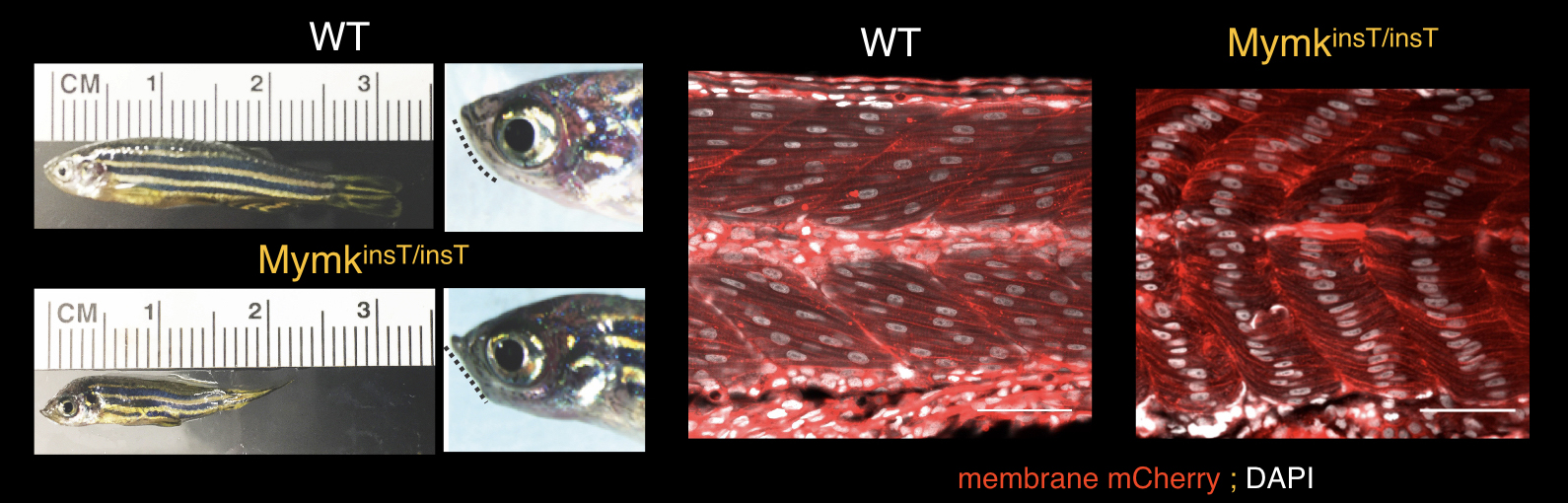
Figure 5. Zebrafish model of Carey-Fineman-Ziter syndrome resulting from loss of MYMK function. A 6-month-old male wildtype zebrafish (left top) compared to an age- and sex-matched Mymk loss-of-function fish (MymkinsT/insT, left bottom). Male and female mutant zebrafish are small and have a flattened/retrognathic jaw (indicated by dotted line) not appreciated during larval and early juvenile stages. By 3 months of age, jaw weakness prohibits the mutant zebrafish from fully closing their mouths. Middle, right: 48 h.p.f. embryos stained with membrane-bound mCherry (red) and DAPI (white) to label the forming myofibers and nuclei, respectively. Wildtype embryo myofibers are multinucleated, while mymkinsT/insT embryo myofibers elongate and differentiate, but fail to fuse. From Di Gioia et al, Nature Commun, 2017. PMCID: PMC5504296 >
Disease Modeling & Mechanism
The Engle Lab models CCDDs primarily in Mus musculus (mouse) and Danio rerio (zebrafish) as well as in vitro, in yeast, and in embryonic stem cells. Cranial nerve anatomy and function is conserved between zebrafish, mouse, and humans, and both fish and mice have proved to be excellent species in which to model CCDDs.
Mouse (and in vitro) Modeling of CCDDs
Starting with BAC recombineering and now using CRISPR-Cas9 technologies, the lab has successfully modeled multiple forms of congenital fibrosis of the extraocular muscles (CFEOM) and Duane retraction syndrome in mice. We use fluorescent reporter mice and have developed imaging approaches to visualize the normal and abnormal development of cranial motor nerves in three dimensions.
Each mouse model has recapitulated the human phenotype and permitted the neurodevelopmental definition of disease mechanism. We have found that CCDDs can arise from failure of motor neuron development (e.g. HOXA1, HOXB1, PHOX2A, MAFB, SALL4). As a result, the motor neurons are respecified or die, and the target muscle(s) are not innervated. We have also found that CCDDs can arise from failure of cranial motor axon growth and guidance (e.g. KIF21A, TUBB3, TUBB2B, CHN1). In these cases, the motor neuron has the correct identity but its axon either stalls or is misguided. Remarkably, in many cases if misguided cranial motor axons reach the incorrect extraocular or facial muscle both the muscle and axon will survive. This results in aberrant patterns of eye or face movements that are referred to as synkinesis. Synkinesis is key to the diagnosis of Duane retraction syndrome and is also found in many other CCDDs.
Our mechanistic studies have revealed that dominant missense mutations in KIF21A and CHN1 attenuate the autoinhibition of the encoded proteins, the anterograde kinesin KIF21A and the Rac-GAP alpha-2-chimaerin, respectively. Attenuation of KIF21A results in loss of temporal and spatial control of the activity of this widely-expressed kinesin, causing stalling of a subset of axons in the oculomotor nerve in the mouse model. Attenuation of alpha-2-chimaerin results in hyperactivity of its Rac-GAP activity and lowering of Rac levels in developing neurons, resulting in stalling of the abducens nerve in the mouse model. Our mechanistic studies have revealed that dominant missense mutations in TUBB3 and TUBB2B result in incorporation of the mutant beta-tubulin isoforms into the microtubule, decreasing microtubule dynamics, reducing motor protein trafficking, and resulting in oculomotor and trochlear misguidance in our mouse models.
Zebrafish Modeling of CCDDs
In addition to mice, our lab uses zebrafish to model CCDDs and myopathic phenomimics. These are disorders that mimic CCDDs but result from a primary error in extraocular (e.g. MYF5) or facial (e.g. MYMK) muscle development. We found that MYMK mutations cause Carey-Fineman-Ziter (CFZ) syndrome and successfully modeled CFZ in zebrafish.
We are currently using zebrafish to functionally validate candidate genes derived from exome and genome sequences of human research participants with CCDDs. Using CRISPR-Cas9 genome editing, we are performing a moderate-throughput screen in zebrafish to determine which genes are causal for these phenotypes. We are then conducting targeted mechanistic studies to validate these findings.
Publications
1. Park JG, Tischfield MA², Nugent AA², Cheng L, Di Gioia SA, Chan W-M, Maconachie G, Bosley TM, Summers CG, Hunter DG, Robson CD, Gottlob I, Engle EC*. Loss of MAFB function in human and mouse causes Duane syndrome, aberrant extraocular muscle innervation, and inner ear defects. Am J Hum Genet. 2016 Jun 2;98(6):1220-7. PMCID: PMC4908193 >
2. Nugent AA, Park JG, Wei Y, Tenney AP, Gilette NM, DeLisle MM, Chan W-M, Cheng L, Engle EC*. Mutant alpha2-chimaerin signals via bidrectional ephrin pathways in Duane syndrome. J Clin Invest. 2017 May 1;127(5):1664-1682. PMCID: PMC5409791 >
3. Michalak SM¹, Whitman MC¹, Park JG¹, Tischfield MA, Nguyen EH, Engle EC*. Ocular motor nerve development in the presence and absence of extraocular muscle. Invest Ophthalmol Vis Sci. 2017 Apr 1;58(4):2388-2396. PMCID: PMC5403115 >
4. Di Gioia SA, Connors S², Matsunami N², Cannavino J², Rose MF², Gilette NM, Artoni P, de Macena Sobreira NL, Chan W-M, Webb BD, Robson CD, Cheng L, Van Ryzin C, Ramirez-Martinez A, Mohassel P, Leppert M, Scholand MB, Grunseich C, Ferreira CR, Hartman T, Hayes IM, Morgan T, Markie DM, Fagiolini M, Swift A, Chines PS, Speck-Martins CE, Collins FS, Jabs EW, Bönnemann CG, Olson EN, Moebius Syndrome Research Consortium, Carey JC*, Robertson SP, Manoli I*, Engle EC*. A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nature Commun. 2017 Jul 6;8:16077. PMCID: PMC5504296 >